- IR情報
- 株主・投資家サポート
- よくあるご質問
よくあるご質問
会社に関するご質問
-
会社設立はいつですか?
-
日本オルガノン株式会社をスピンオフし、2003年4月、神戸市に設立。同年10月よりキナーゼに特化した創薬支援事業および創薬事業の展開を目的として業務を開始しました。
-
設立経緯を教えて下さい。
-
当社は、鐘紡から新薬事業の営業譲渡を受けた、オランダの製薬企業Organon N.V.(以下、「オルガノン」)の日本法人、日本オルガンノン株式会社(以下、「日本オルガノン」)からスピンオフしてできた会社です。2002年にオルガノンの主力製品であったうつ病の薬が米国で特許切れとなり、オルガノンが全世界的なリストラを開始しました。その当時、日本オルガノンの医薬研究所の幹部であった創業メンバーは、日本オルガノン時代から特化して研究してきたキナーゼには将来性があると考え、同社の支援を得て独立いたしました。
-
社名の由来を教えて下さい。
-
社名である「カルナ(CARNA)」は、ローマ神話の「人間の健康を守る女神」です。また、「身体の諸器官を働かせる女神」、「人間生活の保護女神」などとも言われています。
当社グループは生命科学「バイオサイエンス(Bioscience)を探求することで、「人々の生命を守り、健康に貢献することを目指す。」ことを基本理念としています。まさに「カルナ(CARNA)」でありたいと思っています。 -
上場したのはいつですか?
-
2008年3月25日にジャスダック証券取引所NEO市場(現・東京証券取引所グロース市場)に上場しました。
-
強みを教えて下さい。
-
当社グループは、キナーゼを標的にした創薬研究に必要な材料および情報を提供する日本でも数少ない会社のひとつです。特に、創薬研究に必須であるキナーゼタンパク質は、世界で最も多種類の品揃えを誇り、それらを用いた化合物の網羅的なプロファイリングは、創薬研究に有用な情報を提供することができます。キナーゼタンパク質は、遺伝子の取得からタンパク発現〜精製までを自社で一貫して行っていますので、高品質の製品をお客様のニーズに合わせて幅広くご用意することができます。
これらの世界有数のキナーゼタンパク質創薬基盤技術は、自社の創薬研究にも活用することができます。 -
会計監査人を教えて下さい。
-
有限責任監査法人トーマツを会計監査人として選任しております。
詳しくは「会計監査の状況」をご覧下さい。 -
所在地を教えて下さい。
-
当社へのアクセス(当社の所在地)は「会社概要 > アクセスマップ」をご覧下さい。
経営方針・経営戦略に関するご質問
-
中長期的な経営戦略について教えて下さい。
-
中長期的な経営戦略については「中期経営計画」をご覧下さい。
-
配当政策について教えて下さい。
-
当社は創薬型バイオベンチャーとして研究開発に先行投資をすることをビジネスモデルと事業しておりますが、早期の全社黒字化に向けて取り組んでおります。株主の皆さまへの利益還元は重要な経営課題と認識しており、今後の経営成績及び財務状態を勘案し、利益配当等を含め総合的に検討してまいります。
-
創薬の収益モデルについて教えてください。
-
当社が創製した医薬品候補化合物の知的財産権に基づく開発・商業化の権利を製薬会社等に導出(ライセンスアウト)し、その対価として契約一時金、一定の開発段階を達成した際のマイルストーン、新薬の上市後の売上高に応じたロイヤリティ収入を獲得するビジネスモデルです。
比較的早期に有効性が確認できる「がん領域」は最大フェーズ2試験まで当社で臨床試験を実施し、それ以外の疾患はフェーズ1試験もしくは前臨床試験まで実施し、早期ライセンスアウトすることを基本方針としています。 -
コーポレート・ガバナンスに対する取り組みについて教えて下さい。
-
当社グループは、経営の健全性ならびに透明性を高めることを通じて企業価値の最大化を図ることが重要であると考えております。このため、コーポレート・ガバナンスの強化を重要な経営課題と認識し、業務執行に対する厳正な監督機能の充実や内部統制システムに基づく業務執行の妥当性、違法性ならびに効率性のチェック・管理機能を有効に発揮させることによって、経営の健全性ならびに透明性の向上に積極的に取り組んでおります。さらに役員および従業員のコンプライアンスの徹底についても、重要施策として積極的に取り組んでおります。詳しくは「コーポレート・ガバナンス」をご覧下さい。
-
ビジョンを教えてください。
-
当社グループの大きな発展・飛躍は創薬事業の成否が鍵を握るものと考えております。がん疾患や自己免疫疾患などで苦しむ患者の方々に、一刻も早く有効な治療薬をお届けしたいと考えております。
米国では、バイオベンチャーが育ち、創薬が国を支える産業になっております。当社グループもこれからの発展により、国を支える企業の一員として飛躍したいと考えております。
決算・財務に関するご質問
-
最新の業績教えて下さい。
-
業績の詳細は「決算短信」をご覧下さい。
-
決算関連資料の入手場所を教えて下さい。
-
決算関連資料は「IR資料」をご覧下さい。
-
決算期を教えて下さい。
-
12月31日です。
-
決算発表日はいつですか?
-
決算発表のスケジュールは「IRスケジュール」をご覧下さい。
株式に関するご質問
-
証券コードを教えて下さい。
-
4572です。
-
売買単位株式数を教えて下さい。
-
100株です。
-
株主総会はいつ開催されますか?
-
毎年3月に開催します。
-
株主優待制度はありますか?
-
株主優待の制度はありません。
-
QUOカードはどのような基準で送付されますか?
-
当社では、株主総会における議決権行使促進の一環として、2021年3月開催の株主総会以降、議決権を有効にご行使いただいた株主様(当日ご出席の株主様を含みます)の中から、抽選で300名様にQUOカードを贈呈しております。
QUOカードは、原則として5月下旬頃に郵送にてお送りいたします。なお、抽選および送付業務は外部に委託し、厳正に行っております。
※当選者の発表は、発送をもって代えさせていただきます。 -
2024年10月18日付「第三者割当により割り当てられた株式の譲渡に関する報告書」に関して、以下の内容を教えて下さい。
- 割当先(Athos Asia Event Driven Master Fund)は、譲渡後も、譲渡株式に係る議決権を所有しているかどうか教えて下さい。
- 譲渡株式は空売りに利用されているのではないでしょうか?
- 「6 譲渡の理由」なお書きの「譲渡後においても本株式の経済的損益を引き続き享受している」の意味を教えて下さい。(2024.10)
-
質問内容について、割当先であるAthos Asia Event Driven Master Fund(以下「Athos」)より、以下のコメントを受けとりましたのでお知らせいたします。
- Athosは、本譲渡株式について、譲渡後も譲渡先の許可を得ることで、議決権の行使に関与することが可能です。譲渡先において、特別な理由がない限り、議決権行使への関与について許可される理解です。
- 譲渡先は、本譲渡株式の状況についてAthosに報告を行う義務があり、Athosは、譲渡先に本譲渡株式を空売りに用いられる貸株等の二次利用に利用しないことを要請し、了承いただいています。
- なお、本譲渡は、Athosの投資に関する実務的なルールへの抵触を回避するため、当社株式の持株比率を下げる必要が生じたため実施しましたが、Athosは譲渡後も引き続き本譲渡株式の経済的損益を享受しており、議決権行使に関与することが可能です。
また、「6. 譲渡の理由」のなお書きの「譲渡後においても本株式の経済的損益を引き続き享受している」とは、以下のことを意味します。
Athosは、株式を譲渡した後も、譲渡先との契約において、その株式から生じる利益を得る権利を有し、また、損失が発生した場合にはその損失を負担します。つまり、株式を直接保有していなくても、依然としてその株式から生じる経済的な利益や損失に関与しています。
なお、「第三者割当により割り当てられた株式の譲渡に関する報告書」は、割当先から当社への通知の文言に沿って作成しております。(2024.10)
パイプラインに関するご質問
-
新たに開始されるAS-1763 フェーズ 1b 試験 用量拡大パートの内容を分かりやすく説明してください。(2024.9)
-
用量拡大パートの概要は以下の通りです。試験の詳細は、今後学会で発表いたします。
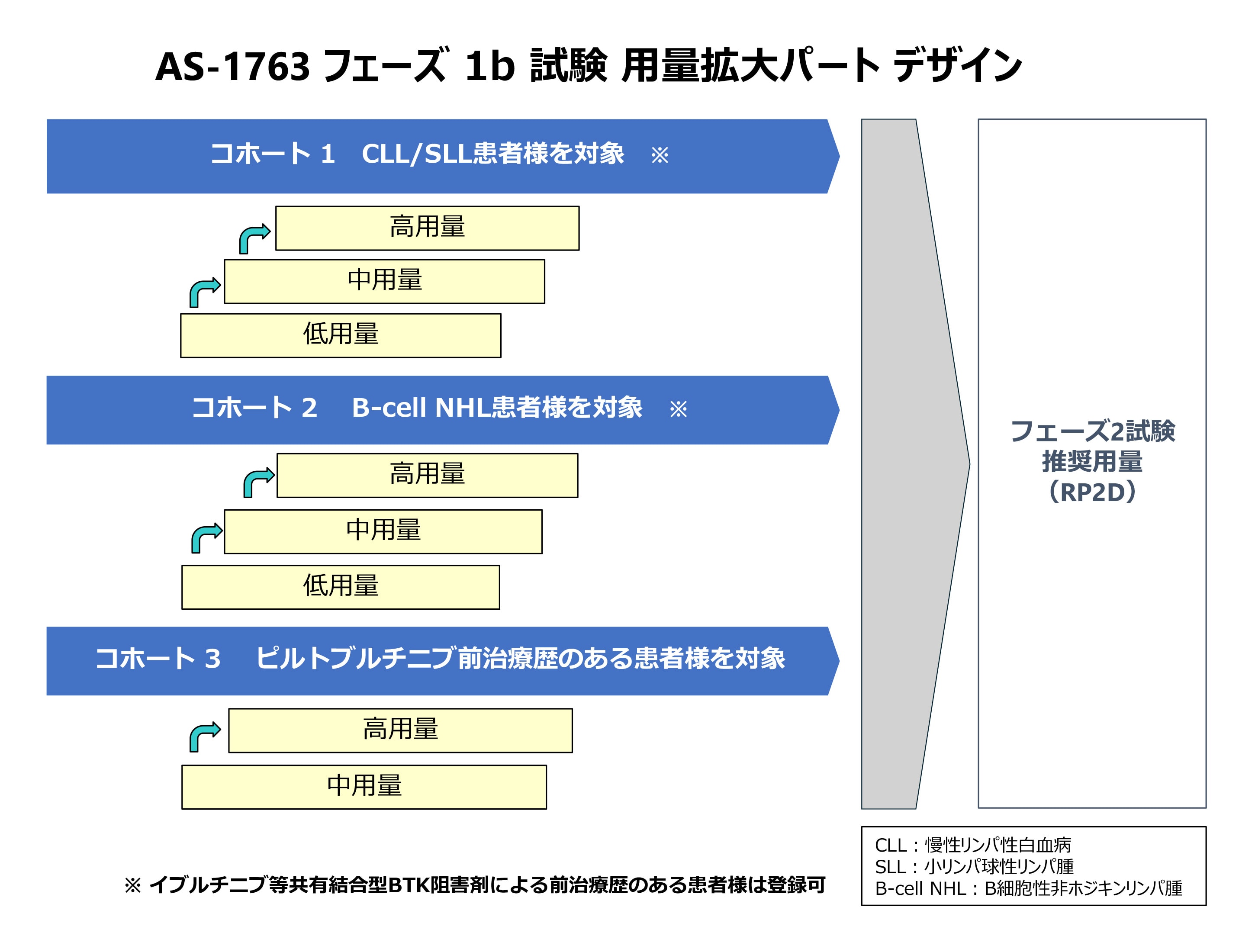 今回の試験は、患者様を3つの群に分けて、AS-1763を投与して有効性と安全性を調べます。コホート1はCLL/SLL患者群、コホート2はB-cell NHL患者群、コホート3はピルトブルチニブ前治療歴のある患者群です。いずれも2ライン以上の全身治療歴のある患者様が対象となります。AS-1763は1日2回経口投与し、原則、2サイクル(1サイクル28日)ごとにCT検査、血液検査等を実施して効果を判定します。副作用で投与が継続できなくなるか、疾患の増悪が認められるまでAS-1763の投与は続きます。各コホートにつき、2用量又は3用量を設定し、各用量群に所定の数の患者様にエントリーいただき、割り当てられた用量のAS-1763を投与します。各用量の効果と安全性を総合的に評価し、次段階のフェーズ2試験の推奨投与量を決めていきます。用量漸増パートでは、用量ごとに慎重に安全性をチェックする期間が必要でしたが、用量拡大パートでは、多施設で同時に患者様のリクルートを進めることができるため、臨床試験は用量漸増パートよりもさらに早く進むことになります。また、この用量拡大パート(コホート3)では、ピルトブルチニブが効かなくなった患者様や副作用で投与を継続できなくなった患者様への投与が実施されるので、ピルトブルチニブとの差別化データが得られることが期待されます。(2024.9)
今回の試験は、患者様を3つの群に分けて、AS-1763を投与して有効性と安全性を調べます。コホート1はCLL/SLL患者群、コホート2はB-cell NHL患者群、コホート3はピルトブルチニブ前治療歴のある患者群です。いずれも2ライン以上の全身治療歴のある患者様が対象となります。AS-1763は1日2回経口投与し、原則、2サイクル(1サイクル28日)ごとにCT検査、血液検査等を実施して効果を判定します。副作用で投与が継続できなくなるか、疾患の増悪が認められるまでAS-1763の投与は続きます。各コホートにつき、2用量又は3用量を設定し、各用量群に所定の数の患者様にエントリーいただき、割り当てられた用量のAS-1763を投与します。各用量の効果と安全性を総合的に評価し、次段階のフェーズ2試験の推奨投与量を決めていきます。用量漸増パートでは、用量ごとに慎重に安全性をチェックする期間が必要でしたが、用量拡大パートでは、多施設で同時に患者様のリクルートを進めることができるため、臨床試験は用量漸増パートよりもさらに早く進むことになります。また、この用量拡大パート(コホート3)では、ピルトブルチニブが効かなくなった患者様や副作用で投与を継続できなくなった患者様への投与が実施されるので、ピルトブルチニブとの差別化データが得られることが期待されます。(2024.9)
-
複数の企業が開発を進めているBTK分解誘導剤(BTK degrader)の臨床試験が順調に進んでいると聞きましたが、BTK分解誘導剤はAS-1763の競合薬となり得るのでしょうか。(2024.8)
-
近年、タンパク質分解誘導剤が新しいメカニズムの創薬として注目されており、BTKを標的としたBTK分解誘導剤の臨床試験も進めれらています。いくつかのBTK分解誘導剤に関しては臨床試験の途中経過が学会等で発表されています。当社のAS-1763は、初期の臨床結果ではありますが、これらのBTK分解誘導剤とほぼ同等の奏効率を示しております。また、現時点におきまして、有害事象の発現頻度並びに重症度は、AS-1763のほうが低く、安全性に優れていると考えています。また、AS-1763の治験に参加いただいている先生方は、BTK阻害薬に精通する非常に著名な方々であり、AS-1763の治験への患者エントリーを積極的に進めていただいていることから、BTK分解誘導剤と比べてAS-1763のもつポテンシャルを高く評価していただいていると考えています。(2024.8)
-
sofnobrutinib (AS-0871)に関して、導出パッケージとはどのようなものでしょうか?また、パートナリング活動について、進捗状況を開示して欲しい。パートナリング活動中は、臨床開発は進捗することなく止まったままでしょうか。(2024.5)
-
sofnobrutinibは、慢性特発性蕁麻疹(chronic spontaneous urticaria、以下CSU)を最初の適応疾患として開発を目指して、オランダでフェーズ1試験を実施いたしました。本フェーズ1試験は、健常人を対象として薬剤の安全性、忍容性および薬物動態プロファイルを確認することが目的であり、当該項目に関して良好な結果が得られています。当該結果の最終報告書を2023年11月に受領し、その内容を基に導出パッケージを作成しました。当該結果に関する報告書及びデータは膨大な量であり、さらに臨床試験のために実施した、多数の非臨床試験の報告書・データも別途存在しております。それらの報告書、データに関して、交渉相手先がdue diligence (DD)において詳細に調査いたします。従いまして、交渉がスムーズに進むように予め、DD用に取りまとめたものを用意しておき、またそこから要点だけを書きだした秘密保持契約下における開示資料、さらに重要な個所だけにした初期面談用資料を作成することを「導出パッケージの作成」と呼んでいます。
通常、パートナリング活動では、興味を有する会社との面談、その後の数回のフォローアップを経て、秘密保持契約を締結した時点から、本格的な交渉が始まりますが、ここまでに数か月を要することが一般的です。そこから詳細な議論、膨大な資料のDD等を経て、契約がまとまるまでにさらに数か月以上を要しますので、できるだけスムーズにDDが開始できるようにパッケージ化することが重要です。
sofnobrutinib (AS-0871)のパートナリング活動の進捗状況につきましては、交渉の状況自体、交渉の駆け引きに重要な情報となるとともに、秘密保持契約では、導出交渉をしている事実も秘密保持の対象となっていることが多いため、その開示について慎重にならざるを得ないことをご理解いただけますと幸いです。
また、通常、医薬品開発においては、臨床試験以外にもフェーズ2試験以降の臨床開発計画の遂行に重要な非臨床試験があり、これらは非常に時間を要することから、臨床試験と並行して実施することが一般的です。sofnobrutinibに関しても、現在、このような非臨床試験を実施しており、パートナリング活動をしながら、臨床試験以外の部分で開発を進めています。(2024.5)
その他のご質問
-
2025年3月25日開催の株主総会及び事業説明会における質疑の内容を教えて下さい。(2025.4.1)
-
株主総会及び事業説明会における質疑の中で、重要なものについてお知らせします。わかりやすくお伝えするため、内容をまとめるとともに、表現を整理、補足しております。(2025.4.1)
(資金調達および継続企業の前提に関する注記について)
質問: 継続企業の前提に関する重要な不確実性は、どうしたら解消されると考えていますか?
回答: 2024年12月末時点の財務諸表において、「継続企業の前提に関する注記」を付すことになった要因は、端的に申し上げると、2024年12月末の当社の保有資金が21億円程度である一方、2025年12月期に計画している当期損失が同程度の21億円であるということが重要なファクターとなりました。
このため、2025年中に創薬事業において大型の導出が実現する等によって多額の売上を計上することができれば、資金繰り上の懸念がなくなるため、重要な不確実性は解消されるものと考えておりますが、導出の実現までに資金繰り上必要があれば、資金調達を検討することになります。
従前は、十分な手元資金を用意しておくという方針の下、資金調達を計画・実施していましたが、今後、マイルストーンが達成される可能性があることや、資金調達についても多くのファンドが支援を表明していること等を勘案し、具体的に資金調達の必要が生じた時に適切に資金調達を実施する方針としました。
質問: 当社の増資の引受先として、製薬企業や事業会社が現実的ではないと考えている理由を教えて下さい。
回答: 当社は、創薬事業における各パイプラインを、日米欧で同時に開発し、新薬を発売できるメガファーマ或いは大型のバイオベンチャーに導出することを目指しています。そのような企業との関係構築を優先する観点から、メガファーマ等への導出に影響が出る可能性ある製薬企業その他の事業会社からの資本受け入れについて、積極的には検討しておりません。一方で、メガファーマが上場バイオベンチャーの株式について、子会社化することなく一部のみ保有する事例は極めて少数であるため、現実にもメガファーマ等からの資本の受入れは容易に実現しない状況にあると考えております。
(docirbrutinib AS-1763 について)
質問: docirbrutinibのフェーズ1b試験の各コホートについて、患者様の組み入れペースの見通しについて教えて下さい。また、組み入れペースが遅いように感じます。
回答: 現在の組み入れペースは昨年と比べて上昇しております。また、治験施設も徐々に増加しているため、今後の組み入れペースはさらに加速していくものと見込んでいます。フェーズ1b試験で最大100例程度の組み入れを目標にしていますが、試験データの収集状況によって、より少ない症例数で、フェーズ2試験の推奨用量を決定し、終了する可能性もあります。
質問: docirbrutinibのプロファイルをみると、フェーズ2試験が終わった段階で、(新薬の販売を開始するための)申請を出せるのではないでしょうか?
回答: 米国の、医薬品承認の迅速審査制度の一つである迅速承認の指定を受けることによって、フェーズ2試験が終わった段階で承認申請をすることができます。現在承認されているpirtobrutinibが薬剤耐性によって効果が失われると他に治療手段がなくなるため、docirbrutinibがpirtobrutinibに対する薬剤耐性をもつ患者様にも効果があること、或いはpirtobrutinibを凌駕する臨床効果が証明できれば、迅速承認に指定される可能性があると考えています。当社としては、迅速審査制度で指定を受けることを目指し、取り組んでまいります。
質問: 次の学会発表は、2025年6月のEHA(ヨーロッパ血液学会)になるのでしょうか?また、学会発表ではコホート3の症例の結果は含まれるのでしょうか?
回答: ご指摘のとおり、2025年6月のEHA(ヨーロッパ血液学会)で発表すべく準備を進めています。学会発表の実施については、発表テーマを応募し、EHAにおいて採択されて初めて公式発表が可能となります。採択の可否が決まり、公表できる段階になりましたら、お知らせしてまいります。
なお、EHAに採択された場合、コホート3のデータについては、現時点でまだ組み入れができていないため、発表に含まれない可能性が高いと考えています。
質問: フェーズ1b試験中の導出の可能性はあるのでしょうか?
回答: 当社としては、フェーズ1b試験中であったとしても、良い条件であれば導出はあり得ると考えています。
(sofnobrutinib AS-0871 について)
質問: 導出の時期、また、導出一時金の金額の見通しを教えて下さい。
回答: sofnobrutinibについては、胚・胎児発生毒性試験において催奇形性が認められなかった点で既存のBTK阻害剤との差別化ポイントが明確となりました。このため、時間をかけてよりよい条件の獲得を目指すという方針の下、導出交渉を進めています。導出時期及び導出金額の見通しについては、相手のあることであり、交渉に要する期間は案件ごとに非常に差があること、また、金額の見通しの公表は導出先との金額交渉上非常に不利に働くため、いずれも回答は差し控えさせていただきますが、当社としては、可及的速やかに、より多額の導出一時金の獲得に努めてまいります。
(次世代のパイプラインについて)
質問: 次世代のパイプラインについて教えて下さい。
回答: 次世代のパイプラインも着実に育っていますが、公表してすぐにドロップするというようなことは避けたいと考えています。そのため、現在は、前臨床試験等を着実に実行し安全性等を含め臨床試験に確実に進むと判断できた時点で公表することを方針としています。
(創薬支援事業について)
質問: 創薬支援事業の売上について、低迷の理由と2025年12月期の見通しを教えて下さい。
回答: 創薬支援事業において売上が低迷した理由は、米国において、バイオ関連企業への資金供給環境が悪く、多くの会社が研究を打ち切った影響が出たと考えています。また、中国においては、米国による中国バイオ企業との取引制限の影響が出ました。現在、このような状況が徐々に解消されていることを確認しており、2025年12月期の売上は、昨年比で増加する見込みです。 -
2025年8月20日開催の個人投資家向けオンライン会社説明会における質疑応答の内容を教えて下さい。(2025.8.26)
-
2025年8月20日開催の個人投資家向けオンライン会社説明会における質疑応答の内容をお知らせします。また、説明会の中でお答えできなかったご質問について、回答を追加しております。
わかりやすくお伝えするため、内容をまとめるとともに、表現を整理、補足しております。
なお、説明パートにおいても、事前にいただいたご質問の一部に対して回答しております。説明パートの録画を掲載しておりますので、ぜひご参照下さい。(2025.8.26)
(ギリアド社導出済創薬プログラム・DGKα阻害剤GS-9911について)
質問: ギリアド社に導出した創薬プログラムから見出されたDGKα阻害剤GS-9911の状況について教えて下さい。
回答: 2025年8月14日にお知らせしました通り、ギリアド社より、GS-9911は、ギリアド社のポートフォリオにおける優先付けに関する決定を反映し、同社の決算発表資料のパイプラインテーブルから除外された旨の連絡を受けております。なお、本導出契約には、一般的な契約解除に関する事前通知条項が含まれていますが、現時点で通知は受けておらず、契約は有効です。今後、契約の定めに従い、GS-9911の状況に関する報告がなされ、GS-9911の開発状況や方針に関して新たな情報が判明次第、速やかにお知らせいたします。
(docirbrutinib AS-1763 について)
質問: docirbrutinibのフェーズ1b試験における各コホートの患者様の組み入れ状況を教えて下さい。
回答: 2025年8月18日現在、docirbrutinibのフェーズ1b試験における各コホートの患者様の組み入れ状況は以下の通りです。
コホート1 : 11名
コホート2 : 7名
コホート3 : 1名
質問: 2025年第2四半期決算説明資料において、docirbrutinibに関する目標として、フェーズ2試験開始を2025年〜2026年と記載がありましたが、現実的に可能でしょうか?
回答: フェーズ2試験は、フェーズ1試験で得られた有効性、安全性、血中薬物濃度などの結果からフェーズ2試験における推奨用量(RP2D)が確定すれば、開始可能となります。現在の進捗状況を踏まえると、CLL(慢性リンパ性白血病)に関して2026年にはRP2Dの確定が見込まれ、同年中にフェーズ2試験を開始可能と考えています。なお、フェーズ1試験において症例数がまだ十分でないがん種については、引き続きフェーズ1試験への患者様の組み入れを継続する可能性が高いと考えています。
質問: docirbrutinibのフェーズ2試験は、自己資金で実施する方針でしょうか?あるいは、導出後に実施する方針でしょうか?
回答: 現時点において、導出後にフェーズ2試験を開始することを想定しています。
質問: コホート2にはいくつかのがん種が含まれますが、効果が見込まれるがん種の患者登録を増やすことは可能でしょうか?
回答: 現時点では、プロトコールに記載されているがん種全ての患者様の登録を継続する予定ですが、ある程度症例が集まった時点で、効果が期待されるがん種を中心に患者様の組み入れを進めることを考えています。
質問: フェーズ1b試験の各コホートは、どの程度の奏効率であれば望ましい結果と言えるのでしょうか?
回答: 患者様の前治療歴や臨床状態などを考慮して総合的に判断するため、現時点では具体的な奏効率を申し上げることはできません。
質問: 欧州血液学会(EHA2025)における発表データによると、CLLの症例で投薬が中止されたケースがごく少数ありますが、その要因を教えて下さい。肝障害が原因でしょうか?
回答: EHAの発表では、17名のCLL患者が登録され、そのうち2名は病勢の進行、2名は同意撤回により治験を中止しました。いずれも、肝障害が原因ではありません。なお、病勢の進行により脱落した2名は、治療効果が期待される用量として拡大パートで選択した用量(300mg、400mg、500mg)よりも低用量(100mgおよび200mg)群の症例です。
(sofnobrutinib AS-0871 について)
質問: 非臨床試験における「催奇形性が認められない」との結果は、どの程度のメリットになりますか?
回答: 企業からは、催奇形性がないという特徴は先行品と差別化できる重要なポイントとして注目していただいています。また、皮膚科領域の教授(ドクター)からも、クリニックでは催奇形性のある薬剤は処方することをためらうため、大きなメリットになるというご意見もいただいています。
質問: sofnobrutinibの上市の時期はいつ頃になりますか?
回答: sofnobrutinibの上市時期については、導出先企業の開発方針や戦略に依存するため、具体的な時期をお伝えすることは難しい状況です。
(monzosertib AS-0141 について)
質問: monzosertibの臨床試験に関して、計画中の血液がんを対象とした他の薬剤との併用試験について、詳細を教えて下さい。
回答: 血液がんを対象とした併用試験として、非臨床試験の結果(2025年5月1日付リリース「CDC7阻害剤monzosertibに関するアメリカ癌学会(AACR)年次総会でのポスター発表概要」参照)に基づき、標準治療であるアザシチジン(DNMT阻害薬)およびベネトクラクス(BCL-2阻害薬)の2剤にmonzosertibを加えた3剤併用試験を検討しています。今後、血液がん領域の専門医と協議を重ね、試験の方針を決定する予定です。
質問: monzosertibのフェーズ1試験に関して、現時点での奏効率を教えて下さい。
回答: 奏効率に関する情報は、現時点で公開しておりませんので、回答は差し控えさせていただきます。
質問: monzosertibについても、早期の導出を目指した方が良いのではないでしょうか?
回答: 初期段階の化合物、特に、first-in-classの化合物を導出する場合には、その臨床的価値を明確にしたうえで交渉を進める方が、より大きな契約につながると考えています。そのため、monzosertibについては、現在計画中の他の薬剤との併用試験において良好なデータが得られるなど、価値最大化を目指した臨床試験で重要なデータが得られた段階で、導出交渉を進めることを考えています。
(住友ファーマ社との共同研究について)
質問: 住友ファーマ社との共同研究に関して、がん領域についてはカルナバイオが権利を有するということですが、住友ファーマ社との共同研究が進まないとがん領域の研究も進まないのでしょうか?
回答: 住友ファーマ社は、本共同研究により見出されたキナーゼ阻害剤のうち、同社が事業化を進めると判断したものについて、がんを除く全疾患を対象に、臨床開発および販売を全世界で独占的に実施する権利を有しています。一方、がん領域については、当社が権利を有しており、住友ファーマ社との共同研究の進捗にかかわらず、当社の判断で独自に研究を進めることが可能です。
(導出交渉について)
質問: docirbrutinibおよびsofnobrutinibの導出交渉の進捗状況について教えて下さい。「パートナーの確保」とはどのような意味でしょうか?
回答: 導出交渉の進捗状況の公表については、交渉の進展に悪影響を及ぼす可能性があるほか、金額等の条件交渉において非常に不利に働くおそれがあるため、差し控えさせていただいております。当社としては、導出契約の締結の実現に向け、全力で取り組んでおります。
「パートナーの確保」とは、導出契約の締結、あるいは、共同開発先との提携を指します。
(研究開発資金の確保および継続企業の前提に関する注記について)
質問: 来年(2026年)に必要な研究開発資金の確保はどのように行う計画ですか?また、継続企業の前提に関する重要な不確実性解消への施策について教えて下さい。
回答: 創薬事業において、大型の導出が実現し収入を得られた場合には、当該収入を2026年の研究開発資金に充当いたします。一方、導出の実現までに資金繰り上必要となった場合、臨床試験を滞らせないために、新たな資金調達の検討を進めてまいります。資金調達のスキーム、規模については、その時点の状況を踏まえ、最適・最善の方法を選択してまいります。
また、「継続企業の前提に関する注記」につきましては、大型導出が実現することなどにより十分な資金が確保されることで、資金繰り上の懸念が払しょくされ、重要な不確実性は解消されるものと考えております。
(その他)
質問: 探索・研究段階のパイプラインについても、パイプラインテーブルに掲載して欲しい。また、マラリア治療薬の研究開発は、継続していますか?
回答: 探索・研究段階のパイプラインにつきましては、その公表が競合他社への刺激となり、開発スピードを加速させるなどのリスクがあるほか、当該段階では研究継続の不確実性が高く、短期間で中止となる可能性も十分に考えられます。このため、当社では、探索・研究段階のパイプラインについて、パイプラインテーブルへの掲載は行わない方針としております。
なお、マラリア治療薬の研究開発については、現在も継続中です。 -
2026年3月25日開催の株主総会及び事業説明会における質疑の内容を教えて下さい。(2026.4.1)
-
株主総会及び事業説明会における質疑の中で、重要なものについてお知らせします。わかりやすくお伝えするため、内容、表現をまとめ、整理するとともに、補足しております。(2026.4.1)
(docirbrutinib AS-1763 について)
質問: ASH2025で発表したポスターそのものを開示できないでしょうか。
回答: ASHなどの大きな学会においては、有償での参加登録が要件とされており、参加登録を行った方のみが、学会内で発表されるポスターを閲覧する権利を有しています。また、学会発表内容をウエブサイト等に掲載することは論文投稿と同等に扱われるため、将来、論文投稿した際に二重投稿とみなされ、論文が受理されないおそれがあります。さらに、学会で発表したポスターの著作権は、その学会が独占的に利用できるため、一般公開は制限されます。
以上の理由から、当該ポスターをそのまま開示することは困難ですので、著作権等に問題のない範囲で、説明資料を作成し、情報提供を実施しています。
今後も引き続き、ビジュアル資料を追加する等して内容を分かりやすくお伝えできるよう努めてまいります。
質問: ASH2025におけるdocirbrutinibフェーズ1b試験に関する発表について、開示された臨床試験データによれば、MCL の症例数は5症例である一方、ウォーターフォールプロットには MCL の症例が2症例のみ掲載されています。残りの3症例については、コホート3に該当するため、開示されなかったものと理解しています。
そこで、このコホート3に該当する3症例について、当時開示されなかった理由、または現時点において公表可能な情報があれば、ご教示ください。
回答: 各症例について、少なくとも治験薬投与開始後のCT画像データが取得されていなければ、腫瘍縮小率を算出することができず、ウォーターフォールプロットに含めることができません。ご指摘の3症例ついては、患者登録後間もない段階で、初回投与後のCT画像データが未取得であったことから、ウォーターフォールプロットから除外されています。
また、コホート3は、ピルトブルチニブが投与され、かつ治療効果が認められなくなった患者を対象とするコホートです。しかしながら、同剤はCLLについては2023年12月に承認されたばかりであるため、コホート3にエントリー可能な症例は限られており、現状、評価可能なデータを呈示できる段階には至っておりません。
なお、現時点でのコホート3へのエントリー数は、MCL 2例および CLL 2例の計4例です。ピルトブルチニブは徐々に普及してきており、今後、コホート3へのエントリーが進むことを期待しています。
質問: docirbrutinibフェーズ1b試験に関して、当初の計画に照らし、臨床試験のエントリーのスピードが遅いように思います。また、docirbrutinib について、2026年中のフェーズ2試験開始及び導出を目標としていますが、状況を教えてください。
回答: docirbrutinibフェーズ1b試験については、現在、より多くの症例を集積することで信頼性の高いデータを蓄積することを目指しており、最近では長期にコントロールできる症例も増加し、興味深い知見が得られています。現在、どのように進めるのがベストか、治験主導医師など様々な専門家と相談していますが、現時点で順調に進捗していることから、本年中にフェーズ2試験のプロトコールを策定できる可能性が高いと考えています。また、フェーズ2試験は導出先に実施してもらうことが望ましいと考えており、本年中の導出を目指しております。
なお、標準治療薬が存在しない患者の方に対し、実際にdocirbrutinibを投与し、その有効性が確認されるたびに、ご家族を含め、多くの方々に非常に喜んでいただいております。
(発行可能株式総数を増加することについて)
質問: 発行可能株式総数を3000万株から7600万株まで大幅に増やす理由について具体的に説明してください。
回答: 当社は、バイオベンチャーの中でも発行株式数が極めて少ない水準にあり、株式分割を除き、設立以来一度も発行可能株式総数を増加させたことがありません。株式は単なる資金調達手段にとどまらず、成長過程におけるM&A、特に株式交換による技術やパイプラインの獲得において、重要な役割を果たすものです。そのため、将来的な成長戦略を見据え、必要な株式を発行できる体制をあらかじめ整えておくことが望ましいと考えています。
なお、当社といたしましては、バイオベンチャーのビジネスモデルとして、先行投資を通じて、資金調達に伴う株式の希薄化を上回る企業価値を創出することが重要であると考えています。
(その他)
質問: 昨年、CMO(チーフメディカルオフィサー)を設置されましたが、どのような変化を意図され、現実にどのようなオペレーションを担当されているのかお聞かせください。
回答: 当社では、臨床開発体制のさらなる強化を目的として、昨年、CMO(チーフメディカルオフィサー)を設置いたしました。当社のCMOは、外科医としての臨床経験に加え、バイオベンチャーやグローバル製薬企業において、がん領域を中心に、新薬の開発から市場拡大戦略の策定まで幅広い経験を有する人材です。
こうした背景を踏まえ、当社では、開発戦略策定、治験プロトコールの作成、有効性及び安全性の評価など様々な場面において、医学的な助言を行っています。また、臨床開発チームの一員として、米国医師と直接相談しながら医師主導試験を精力的に推進しています。
質問: IRにおいて、一般の投資家にも分かりやすく、インパクトのある表現とすることはできないでしょうか。
回答: IRでは、データがまだ不確実な段階で過度に強い表現を用いると、信用を損なうおそれがあるため、適切な情報発信を行うことを基本方針としています。
従前のIRにおける表現について、控えめな面があったことを認識していますが、現在は、評価すべき点については明確に伝えるよう心掛けており、今後も適切に取り組んでまいります。
質問: インターネット上の掲示板において、当社および当社関係者への誹謗中傷が見受けられますが、対応方針についてお聞かせください。
回答: 当社といたしましても、事実に基づかない誹謗中傷が書き込まれることは、極めて遺憾であり、当社の企業価値向上の観点からも重大な問題であると認識しております。掲示板が健全な議論の場となることを望んでおり、法的措置を含めた適切な対応についての検討を継続してまいります。 -
2026年夏の個人投資家向け会社説明会の開催予定について教えてください。(2026.7.30)
-
毎年夏に開催しております個人投資家向け会社説明会につきましては、2026年は代表取締役社長および臨床開発責任者が登壇する、より充実した内容での開催を予定しております。臨床開発責任者の一時帰国に合わせ、2026年10月の開催を予定しております。詳細が決まり次第、当社ホームページにてお知らせいたします。
- IR資料
- 株式情報
- 株主・投資家サポート
